
林峻宇助理教授研究團隊發表研究成果於Brief Bioinform.
連結網址:https://pubmed.ncbi.nlm.nih.gov/36719112/
Abstract
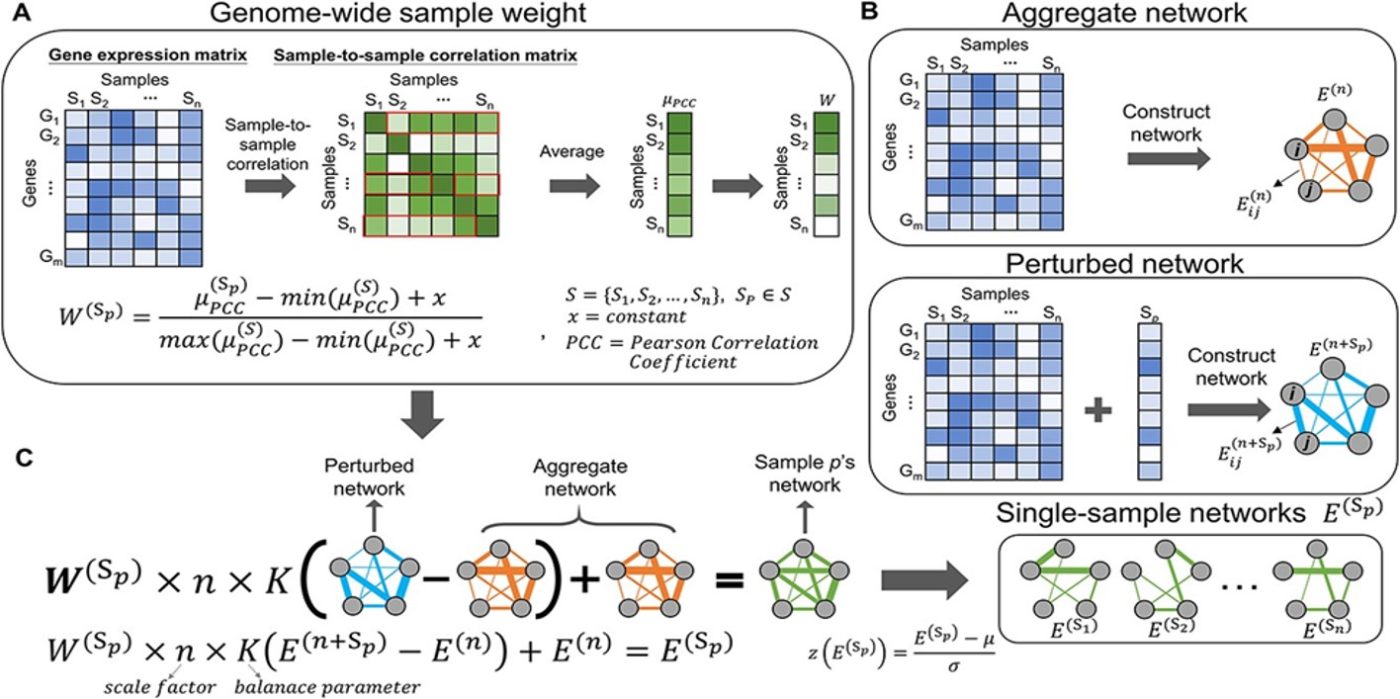
Recently, extracting inherent biological system information (e.g. cellular networks) from genome-wide expression profiles for developing personalized diagnostic and therapeutic strategies has become increasingly important. However, accurately constructing single-sample networks (SINs) to capture individual characteristics and heterogeneity in disease remains challenging. Here, we propose a sample-specific-weighted correlation network (SWEET) method to model SINs by integrating the genome-wide sample-to-sample correlation (i.e. sample weights) with the differential network between perturbed and aggregate networks. For a group of samples, the genome-wide sample weights can be assessed without prior knowledge of intrinsic subpopulations to address the network edge number bias caused by sample size differences. Compared with the state-of-the-art SIN inference methods, the SWEET SINs in 16 cancers more likely fit the scale-free property, display higher overlap with the human interactomes and perform better in identifying three types of cancer-related genes. Moreover, integrating SWEET SINs with a network proximity measure facilitates characterizing individual features and therapy in diseases, such as somatic mutation, mut-driver and essential genes. Biological experiments further validated two candidate repurposable drugs, albendazole for head and neck squamous cell carcinoma (HNSCC) and lung adenocarcinoma (LUAD) and encorafenib for HNSCC. By applying SWEET, we also identified two possible LUAD subtypes that exhibit distinct clinical features and molecular mechanisms. Overall, the SWEET method complements current SIN inference and analysis methods and presents a view of biological systems at the network level to offer numerous clues for further investigation and clinical translation in network medicine and precision medicine.